
Biochemistry: Enzyme Deficiency
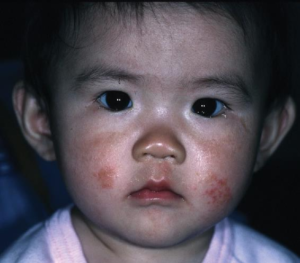
A 6-month-old boy is brought to the office by his mother over concerns of a developmental delay and foul-smelling wet diapers. Pregnancy and birth history are unremarkable. The baby and his mother immigrated to the United States from Russia 1 month ago. On physical examination the patient has a social smile but does not hold his head up on his own or make babbling noises. Brisk reflexes are noted in the upper and lower extremities. Skin examination is shown.
Which of the following enzymes is most likely deficient in this patient?
Important Clues
Let’s show you what we think are the important clues in this vignette and lead-in. We have a much younger age, a six month old boy, and we were told right off the bat certain concerns that made the mom bring the baby in. Developmental delay and foul smelling wet diapers. The stem then gives us an important social history detail: anytime you have a patient and they talk about immigration or where they came from, always make note of that.
To make note of that they also gave us some important physical exam findings as well. It’s definitely important to take note of that, especially when they’re abnormal and then they show us an image of the skin of the six month old boy. Take note of what you’re appreciating in that image.
How Many Steps Does This Question Require?
So then they ask us about an enzyme that is most likely deficient in the patient, so we have a couple steps here.
1) We’ve got to figure out what’s going on with his baby, what’s the condition?
2) What is the enzyme deficiency in that condition?
This looks like a nice, 2 step question.
Let’s go ahead take a look at those answer choices:
A. Branched-chain ?-ketoacid dehydrogenase
B. Dihydrobiopterin reductase
C. Dopamine ?-hydroxylase
D. Homogentisate oxidase
E. Tyrosine hydroxylase
What do you think is the correct answer? Check your answer in the video or scroll down.
.
.
.
.
.
.
.
.
.
.
===
Correct Answer
Answer: B – Dihydrobiopterin reductase
It’s definitely a tough question, so what exactly is going on? What’s the first step? What’s the diagnosis here? This patient has some immigration history, developmental delay, foul smelling odor, and an eczematous rash. If you look at those cheeks what you see is ill-defined erythematous, somewhat scaly rash on the cheeks. Especially in infants, eczema is pretty common on the cheeks. As we get older, it starts to move more to the extremities.
So we have this eczematous rash and these other signs and symptoms, which are concerning for phenylketonuria (PKU). The reason they give us his immigration history is because in the United States, this is usually something that’s discovered at birth when you do your routine newborn screening. It’s likely they wouldn’t not have missed that had they been born in the U.S. However, since they immigrated here from a different country, it’s possible it may have been missed.
PKU is most commonly caused by an enzyme deficiency of phenylalanine hydroxylase (PAH), the first enzyme in the breakdown pathway for phenylalanine. PAH converts phenylalanine to tyrosine, and then tyrosine hydroxylase converts tyrosine to dihydroxyphenylalanine. Both of these enzymes require a cofactor called tetrahydrobiopterin (BH4), which is regenerated by the enzyme dihydropteridine reductase (DHPR). An absence of DHPR yields even more devastating consequences. This disorder is known as “malignant PKU.” Both deficiencies cause a buildup of phenylalanine, and urinary excretion of the excess phenylalanine causes the musty odor characteristic of the disease.
For a quick refresher on amino acid metabolism, check out the Rx Brick on the topic.
Rationales for the Incorrect Answer Choices
So if we go back to our question and look at the other answer choice as well hopefully those clues are now starting to fit.
A. Branched-chain ?-ketoacid dehydrogenase: A deficiency of branched-chain ?-ketoacid dehydrogenase is associated with maple syrup urine disease. This autosomal recessive disorder is caused by an inability to break down the branched-chain amino acids (leucine, isoleucine, and valine). Patients classically present in the first days of life with irritability, vomiting, lethargy, dystonia, and maple syrup smell to urine. This was probably the closest answer choice, but the other clinical signs and symptoms did not really fit with that, as well as the age of presentation for the best answer choice here is B after picking up that this was PKU.
C. Dopamine ?-hydroxylase: The enzyme dopamine ?-hydroxylase is involved in the biosynthesis of norepinephrine and epinephrine, specifically in the step of converting dopamine to norepinephrine (which later becomes epinephrine). A deficiency in this enzyme would present with severe hypotension, hypothermia, or hypoglycemia.
D. Homogentisate oxidase: Alkaptonuria is an autosomal recessive disorder of tyrosine metabolism due to deficient activity of homogentisate oxidase. The disease leads to arthralgias later in life, but the primary sign in an infant would be urine that darkens to black after several hours.
E. Tyrosine hydroxylase: Tyrosine hydroxylase deficiency is associated with oculocutaneous albinism due to an inability to produce melanin from tyrosine. Affected individuals have white hair, blue eyes, and a predisposition toward skin cancer. Suspicion for this disorder is usually aroused based on physical appearance or ophthalmologic exam (which detects visual acuity, photophobia, and nystagmus).


